
Farmakokinetik er den gren af farmakologien der beskæftiger sig med hvad kroppen gør ved lægemidlet – i modsætning til farmakodynamik der beskæftiger sig med hvad lægemidlet gør ved kroppen. Det vil sige farmakokinetik handler om administrationen, distributionen, metabolismen og eliminationen af lægemidler.
Diffusion
Når lægemidler i kroppen skal flytte sig over længere afstande sker det ved bulk flow, dvs. i hold – f.eks. i peristaltiske bølger i tarmene. Når de skal flytte sig over kortere afstande, sker det ved brownske bevægelser (tilfældige bevægelser) der betyder at lægemidlet diffunderer fra et område med høj koncentration til et område med lav koncentration. Denne diffusion sker inden for kompartments (væskefyldte rum som f.eks. ECV eller ICV). Når et lægemiddel skal fra et kompartment til et andet (f.eks. fra ECV til ICV) skal det passere en cellemembran og her kan transporten ske ved passiv diffusion, diffusion gennem vandige porer, fasciliteret diffusion ved carriers, aktiv transport og pinocytose. Den vigtigste mekanisme for lægemidler er passiv diffusion.
Den drivende kraft for diffusion er koncentrationsgradienten. Her kan man beskrive diffusionshastigheden 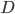 som en funktion af permeabilitetskoefficienten
som en funktion af permeabilitetskoefficienten 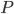 , membranarealet
, membranarealet 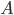 og koncentrationsgradienten
og koncentrationsgradienten 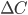 .
.
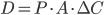
Permeabilitetskoefficienten er afhængig af membrantykkelsen, diffusionskoefficienten af lægemidlet opløst i membranen og lægemidlets opløselighed i lipid. Diffusionskoefficienten afhænger af molekylets størrelse, men denne forskel mellem lægemidler er negligibel og det betyder at diffusionen af et lægemiddel gennem en membran først og fremmest afhænger af lægemidlets lipidopløselighed.
Polariteten af lægemidler
Lipidopløseligheden kan man beskrive ved en fordelingskoefficient, hvor man har taget opløseligheden af stoffet i en apolær væske og divideret med opløseligheden i vand. Det betyder at lipofile lægemidler har en høj fordelingskoefficient og lipidopløselighed – mens hydrofile lægemidler har en lav fordelingskoefficient og lipidopløselighed.
Hydrofile lægemidler har svært ved at blive absorberet, komme ind i celler, passere blod-hjerne barrieren og passere placentabarrieren. Til gengæld har de nemmere ved at blive udskilt i nyrerne og transporteres frit i blodet. Lipofile lægemidler kan omvendt nemt absorberes, komme ind i celler, passere forskellige barrierer – men har sværere ved at blive udskilt med nyrerne og transporteres frit i blodet.
Det betyder at der må være nogle mekanismer i kroppen, som ændrer på polariteten af lægemidler for enten at fremme deres transport, deres metabolisme og deres evne til at blive optaget i forskellige væv. Nogle af disse er vandporer mellem celler, hvor hydrofile lægemidler kan passere, proteintransportere der kan transportere hydrofile molekyler over membraner, proteiner der kan transportere lipider rundt i blodet m.m. Herudover sker der ofte ved metabolisme af lægemidler i leveren en omdannelse der ændrer deres polaritet.
Dette kan også illustreres ved forskellen mellem morfin og heroin:
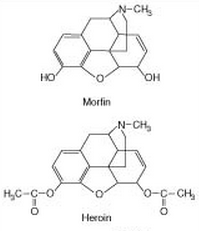
Her kan man se at den eneste forskel mellem de to molekyler er at heroin har acetyleret sine to hydroxygrupper. Dette øger lipidopløseligheden og det er det der gør at heroin nemmere passerer blodhjernebarrieren end morfin. Hvis man injekterede heroin direkte ind i hjernen vil man dog finde ud af at det faktisk ikke har nogen virkning. Dette skyldes at en af de essentielle dele af opoider som er vigtig for binding til receptoren er den hydroxygruppe der findes til venstre på morfin (den højre hydroxygruppe er ikke essentiel for binding). Heldigvis bliver den venstre acetylgruppe på venstre side af heroin hydrolyseret i leveren, så den bliver til en hydroxygruppe igen. Derfor kan heroin aktivere de opoide receptorer, men samtidig være mere lipidopløselig end morfin.
Lægemidler og pH
Ofte er lægemidler også enten syrer eller baser. En syre er et stof hvor der let kan afgives en proton (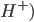 , hvorimod en base er stof hvor der let kan optages en proton (
, hvorimod en base er stof hvor der let kan optages en proton (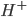 ). En base vil ofte kræve at der er et frit lonepair, hvor elektroner kan doneres til en proton. Syrer og baser kan have forskellig henholdsvis syrestyrke og basestyrke, hvilket henholdsvis betegner deres evne til at afgive eller optage protoner. En stærk syre har således let ved at afgive protoner og en stærk base har let ved at optage protoner.
). En base vil ofte kræve at der er et frit lonepair, hvor elektroner kan doneres til en proton. Syrer og baser kan have forskellig henholdsvis syrestyrke og basestyrke, hvilket henholdsvis betegner deres evne til at afgive eller optage protoner. En stærk syre har således let ved at afgive protoner og en stærk base har let ved at optage protoner.
pH er et mål for surheden af en opløsning (f.eks. vand eller blod) – ikke et mål for styrken af en syre. Det er et mål for hvor mange protoner der har bundet sig til vand i hydroxoniumioner 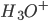 , idet pH defineres som:
, idet pH defineres som:
![pH=-log[H_3O^+]](http://medicin.wiki/wp-content/uploads/2014/09/tex_20a814b79d44f6318426580996450f1b.png)
I og med at syrer og baser kan protoneres og deprotoneres, betyder dette at de kan ændre polaritet, da en protonering af et neutralt molekyle gør molekylet ladet og meget mere polært. Det ladet molekyle har derfor væsentlig større polaritet end det neutrale molekyle. Der vil ofte være en blanding af den depronerede form [deprot] og den protonerede form [prot] i en opløsning. Denne blanding og fordeling mellem de to former vil være direkte afhængig af pH, da lav pH fremmer protonering og høj pH fremmer deprotonering. Omvendt er pH også afhængig af den fordeling der er imellem de to former. Fordelingen kan beskrives ved [deprot]/[prot]. Sammenhængen mellem pH og dette forhold kan beskrives ved pufferligningen eller Henderson-Hasselbach ligningen:
![pH = pK_a + log frac{[deprot]}{[prot]}](http://medicin.wiki/wp-content/uploads/2014/09/tex_dc694e576738a14d0da04ef4e575e022.png)
Af denne formel fremgår også at den 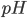 hvor der er lige meget af begge former er når
hvor der er lige meget af begge former er når 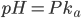 (fordi der er log(1)=0). Vi kan også se at hvis er lavere end
(fordi der er log(1)=0). Vi kan også se at hvis er lavere end 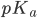 er der mest af den protonerede form og omvendt.
er der mest af den protonerede form og omvendt.
P-glykoprotein
P-glykoprotein er et aktivt transportprotein (en effluxtransporter) der sidder i cellemembranen ved nogle barrierer og aktivt pumper lægemidler og andre stoffer der er fremmede for kroppen (xenobiotika) tilbage hvor de kom fra. Man siger også at p-glykoprotein er en vandskyende støvsuger, der fjerner sine substrater fra cellemembranen.
P-glykoprotein findes bl.a. i følgende væv:
- Enterocytter, hvor de pumper lægemidler tilbage til tarmlumen og modvirker deres optagelse.
- Cancerceller, hvor de forhindrer optagelsen af cytostatica og giver derfor multi-resistens.
- Hepatocytter og galdegangsepithelceller, hvor de øger biliær sekretion af lægemidler.
- Nyretubulusceller, hvor de øger renal udskillelse af lægemidler.
- Endothelceller i hjernekapillærer og epithelceller i plexus choroideus, hvilket nedsætter passagen af lægemidler over blodhjernebarrieren.
- Endothelceller i testiskapillærer, hvilket nedsætter passagen af lægemidler over blodtestisbarrieren.
- Syncototrofoblastceller, hvilket nedsætter passagen af lægemidler over placenta.
Formålet med P-glykoprotein er at beskytte kroppen mod udefrakommende potentielt toksiske stoffer.
Hvis to lægemidler som begge er substrat for P-glykoprotein indtages, kan de udkonkurrere hinanden, hvilket fører til en øget plasmakoncentration af taberen, pga. øget optagelse i tarmene og formindsket udskillelse med nyrerne.
Ophobning af lægemidler i fedtvæv
Nogle lipidopløselige lægemidler kan ophobes i det adipøse væv, hvor de kan udgøre et reservoir hvorfra der langsomt løbende frigives lægemiddel. Dette kan betyde at den terapeutiske virkning af lægemidler kan fortsætte efter man er stoppet med at indtage lægemidlet.
Administration
Administration af et lægemiddel vil sige tilførsel, indgift, applikation og indtagelse af et lægemiddel. Det er altså den måde man tager lægemidlet. Generelt optages et lipidopløseligt lægemiddel hurtigere end et vandopløseligt. De væsentligste administrationsformer er:
- Peroral
- Rektalt
- Intravenøs
- Transdermal
- Intramuskulær
- Subkutant
- Inhalation
- Intraspinalt
- Intratekalt
- Sublingvalt
Peroral administration
Ved peroral indtagelse vil lægemidlet typisk blive absorberet/optaget i tyndtarmen. Her vil lipofile lægemidler blive optaget ved passiv diffusion, hvorimod hydrofile lægemidler stort set ikke absorberes. Fordelen ved peroral administration er helt sikkert at det er en let administrationsvej for patienten. Men der er en del forhindringer, hvoraf disse er nogle af dem:
- Polære lægemidler kan ofte ikke anvende denne administrationsform.
- Forsinket ventrikeltømning kan også forsinke absorptionen.
- P-glykoprotein i enterocytter kan modvirke absorption.
- Kan ikke anvendes til f.eks. bevidstløse patienter.
- Kan ikke anvendes ved kvalme.
- Lægemidler kan destrueres af ventriklens sure pH eller fordøjelsesenzymer i tarmene.
- Kan interagere med andre lægemidler eller fødeemner (kan f.eks. konkurrere med aminosyretransporter).
- Lægemidler kan blive metaboliseret af tarmbakterier eller i tarmepithelet.
- Lægemidler kan fremkalde kvalme ved at irritere slimhinden.
Sublingval administration
Administration under tungen. Her kan meget lipofile lægemidler optages, f.eks. nitroglycerin og nikotin. Fordelen er at man undgår førstegangsmetabolisme ved leveren, fordi det venøse tilbageløb ikke sker til v. portae.
Rektal administration
Her kan meget lipofile lægemidler optages. Fordelen er at man undgår førstegangsmetabolisme af leveren, fordi det venøse tilbageløb ikke sker til v. portae.
Parenteral administration
Administration bl.a. intravenøst, subkutant eller intramuskulært. Her kan hydrofile lægemidler let administreres, da blodet er hydrofilt. Fordelen er også en præcis og hurtig virkning. Ulempen er højere risiko for toksiske virkninger og at det er en besværlig administrationsvej.
Kinetik
Kinetik betyder bevægelse og handler om hastigheden hvormed processer foregår. En førsteordenskinetik betyder at hastigheden v er ligefrem proportional med koncentrationen af lægemidlet [S]. De fleste lægemidler udvikles med førsteordenskinetik:
![v = k cdot [S]](http://medicin.wiki/wp-content/uploads/2014/09/tex_058ab547bf21f3d1f1526f9756e00aa5.png)
En nulteordenskinetik betyder derimod at hastigheden er konstant, uanset koncentrationen af lægemidlet.
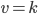
De fleste lægemidlers absorption, distribution og passiv reabsorption i nyretubuli foregår ved førsteordenskinetik. Derimod vil aktiv transport eller enzymmedierede processer være af begrænset kapacitet og kan derfor beskrives ved Michaelis-Menten ligningen:
![v = frac{V_{max} cdot [S]}{K_m + [S]}](http://medicin.wiki/wp-content/uploads/2014/09/tex_40adf40bc368af793e04b13135e088f2.png)
Hvor 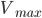 er den maksimale hastighed og
er den maksimale hastighed og 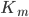 er affiniteten mellem lægemidlet og den aktive transporter, enzymet eller lign.
er affiniteten mellem lægemidlet og den aktive transporter, enzymet eller lign.
Clearence
Når man kigger på eliminationshastigheden 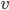 vil denne kunne beskrives som værende ligefrem proportionel med plasmakoncentrationen af lægemidlet
vil denne kunne beskrives som værende ligefrem proportionel med plasmakoncentrationen af lægemidlet 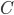 . Clearence
. Clearence 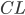 defineres som proportionalitetskonstanten mellem eliminationshastigheden fra et organ eller kroppen (fra plasma) og plasmakoncentrationen.
defineres som proportionalitetskonstanten mellem eliminationshastigheden fra et organ eller kroppen (fra plasma) og plasmakoncentrationen.
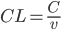
Man kan også tale om en clearence for væv, som f.eks. fra leveren 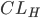 eller nyrerne
eller nyrerne 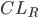 . Den totale clearence kan udtrykkes som summen af disse to, da lægemidler hovedsaligt enten udskilles via nyrerne eller via leveren:
. Den totale clearence kan udtrykkes som summen af disse to, da lægemidler hovedsaligt enten udskilles via nyrerne eller via leveren:
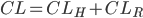
Man kalder den fraktion mellem den del der udskilles via nyrerne og den totale clearence for (f_e):
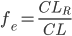
Den fraktion mellem den del der udskilles via leveren og den totale clearence kaldes for 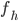 :
:
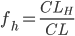
En nedsat nyrefunktion påvirker ikke den hepatiske clearence – og omvendt påvirker en nedsat leverfunktion ikke den renale clearence. Begge tilfælde vil føre til et fald i total clearence.
Ekstraktionsratio
Man definerer en ekstraktionsratio 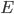 som forskellen mellem den tilførte koncentration af et lægemiddel
som forskellen mellem den tilførte koncentration af et lægemiddel 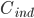 og den fraførte koncentration
og den fraførte koncentration 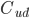 divideret med den tilførte koncentration til et organ. Det antages at der ikke syntetiseres eller bindes noget af lægemidlet i organet.
divideret med den tilførte koncentration til et organ. Det antages at der ikke syntetiseres eller bindes noget af lægemidlet i organet.
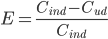
Man kan beskrive clearence som værende lig med flowet til organet 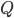 gange med ekstrationsratioen i organet:
gange med ekstrationsratioen i organet:
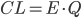
Udfra denne ligning kan man også beskrive clearence som værende det volumen plasma der indeholder den mængde lægemiddel der elimineres pr. tidsenhed. Derfor har clearence også enheden 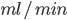 eller
eller 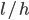 .
.
Hvis man måler plasmakoncentrationen som funktion af tiden efter indtagelse af et lægemiddel, vil koncentrationen først stige til et maksimum og herefter aftage igen. Det giver en kurve der ser nogenlunde således ud:
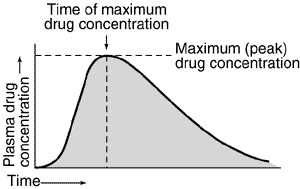
Her kan det vises at man kan bestemme lægemidlets clearence som værende forholdet mellem dosis og arealet under denne kurve, kaldet 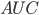 (area under the curve).
(area under the curve).
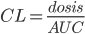
Man inddeler nu lægemidler på baggrund af størrelsen af clearence:
- Lav-clearence lægemidler hvor CL < 18 l/h
- Intermediær-clearence lægemidler hvor 18 l/h < CL < 42 l/h
- Høj-clearence lægemidler hvor CL > 42 l/h
Ved lav eller intermediær clearence lægemidler er CL konstant uanset om man giver lægemidlet peroralt eller intravenøst. Men for høj-clearence lægemidler der metaboliseres i leveren er der forskel, idet CL er større ved peroral administration end ved intravenøst administration pga. førstepassagemetabolisme der reducerer AUC.
Clearence kaldes for en primær farmakokinetisk parameter.
Fordelingsvolumen
Hvis man betragter kroppens samlede indhold af et lægemiddel 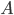 , vil denne også være ligefrem proportional med plasmakoncentrationen . Proportionalitetskonstanten kalder man for fordelingsvolumen
, vil denne også være ligefrem proportional med plasmakoncentrationen . Proportionalitetskonstanten kalder man for fordelingsvolumen 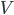 og angives i enheden
og angives i enheden 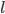 .
.
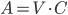
Indholdet af lægemidlet i plasma kan beregnes ved plasmavolumen gange med plasmakoncentrationen og hvis man dividerer dette med fordelingsvolumen får man en fraktion der beskriver hvor meget af lægemidlet i kroppen der findes i plasma. En tommelfingerregel er at et fordelingsvolumen på 2-5 l betyder at stort set alt findes i plasma, mens et fordelingsvolumen på 10-50 l betyder at lægemidlet primært findes enten i ECV eller i kroppens samlede vandfase. Nogle lægemidler kan sagtens have et fordelingsvolumen på næsten 60000 l.
Fordelingsvolumen kaldes for en primær farmakokinetisk parameter.
Man kan ved hjælp af clearence og fordelingsvolumen beregne hvor stor en del af den lægemiddel der findes i kroppen, der elimineres pr. tid:
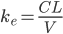
Hvis man betragter et lægemiddel der indgives intravenøst og måler plasmakoncentrationen som funktion af tiden, vil man få samme kurve som længere oppe – bortset fra at man har maks koncentration lige fra starten. Her vil følgende ligning beskrive den eksponentielt aftagende kurve:
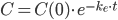
Hvor (C) er plasmakoncentrationen til en bestemt tid og (C(0)) er startkoncentrationen. Hvis man indtegner (ln(C)) som funktion af tiden får man en ret linje med hældning (k_e).
Man kan nu beregne fordelingsvolumen til tiden 0 ved:
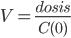
Man kan også beregne fordelingsvolumen udfra dosis, eliminationskonstanten og AUC:
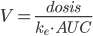
Halveringstiden
Halveringstiden er den tid hvor plasmakoncentrationen af et lægemiddel bliver halveret. Denne kan beregnes ved:
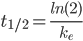
Dette kan også omskrives til:
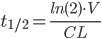
Af dette kan man se at halveringstiden både afhænger af clearence og fordelingsvolumen for lægemidlet. Derfor kaldes det en sekundær farmakokinetisk parameter.
Absorption
Medmindre lægemidlet er lokalt virkende, skal det ind i blodbanen og man kalder overgangen fra administrationen til blodbanen for absorption af lægemidlet. Efter absorptionen opfører lægemidlet sig som om det var givet intravenøst.
Så længe absorptionshastigheden er større end eliminationshastigheden, vil plasmakoncentrationen stige. Når hastighederne er lige store har vi den maskimale plasmakoncentration 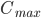 og så falder plasmakoncentrationen herefter. Tidspunktet ved den maksimale plasmakoncentration kaldes for
og så falder plasmakoncentrationen herefter. Tidspunktet ved den maksimale plasmakoncentration kaldes for 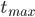 .
.
Biotilgængelighed
Biotilgængelighed betegner både hastigheden hvormed lægemidlet når frem til sit target og den fraktion(F) af lægemidlet der når ind i det systemiske kredsløb. Biotilgængeligheden (hvor man mener fraktionen) kan beregnes ved:
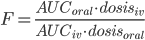
Steady state
Når man indtager et lægemiddel intravenøst, vil man kalde den situation hvor infusionshastigheden er lig med eliminationshastigheden for steady state (en slags ligevægt i hele kroppen mht. lægemidlet). Man kan beregne den plasmakoncentration 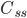 hvormed der er steady state udfra infusionshastigheden
hvormed der er steady state udfra infusionshastigheden 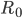 og clearence:
og clearence:
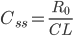
er altså den plasmakoncentration der kan opnås ved gentagen indtagelse af et lægemiddel. Ved infusion vil plasmakoncentrationen være 50% af steady-state koncentrationen efter en halveringstid, 75% af steady-state koncentrationen efter 2 halveringsitder etc. Med andre ord når man aldrig steady-state koncentrationen, men jo længere tid der går jo mere nærmer man sig den. En tommelfingerregel er at man har nået steady-state koncentrationen efter 5 halveringstider.
Bemærk at en ændring i infusionshastighed ikke ændrer på tiden til steady-state men kun koncentrationen. Omvendt vil en ændring af fordelingsvolumen ændre på tiden til steady-state – men ikke på koncentrationen. En ændret clearence (men ikke på dosis og fordelingsvolumen) vil ændre både på tiden og koncentrationen.
Dosis med intervaller
Ofte vil man i praksis give infusioner med intervaller imellem. Her kan gennemsnits steady-state koncentrationen beregnes ved van Rossums formel:
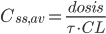
Hvor 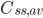 er gennemsnits steady-state koncentrationen og
er gennemsnits steady-state koncentrationen og 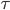 er intervallet mellem hver infusion. Denne ligning er meget vigtig fordi den viser at hvis man giver et lægemiddel med intervaller imellem og ikke en løbende dosis, afhænger plasmakoncentrationen af dosis, intervallet og clearence – ækvivalent med hvis man gav dosis løbende.
er intervallet mellem hver infusion. Denne ligning er meget vigtig fordi den viser at hvis man giver et lægemiddel med intervaller imellem og ikke en løbende dosis, afhænger plasmakoncentrationen af dosis, intervallet og clearence – ækvivalent med hvis man gav dosis løbende.
Hvis man giver dosis med intervaller, vil man se at plasmakoncentrationen flukterer mellem en minimumværdi 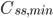 og en maksimumværdi
og en maksimumværdi 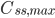 . En tommelfingerregel er så at steady state er nået efter ca. 5 halveringstider. Begrebet fluktation defineres som:
. En tommelfingerregel er så at steady state er nået efter ca. 5 halveringstider. Begrebet fluktation defineres som:
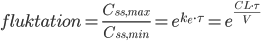
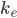 er forholdet mellem clearence og fordelingsvolumen, og derfor fremgår det af ligningen at hvis en person har en lav clearence, pga. en anden enzymaktivitet, så vil fluktationen også formindskes. Man kan også se at fluktationen falder hvis intervallet bliver mindre eller hvis fordelingsvolumen bliver større.
er forholdet mellem clearence og fordelingsvolumen, og derfor fremgår det af ligningen at hvis en person har en lav clearence, pga. en anden enzymaktivitet, så vil fluktationen også formindskes. Man kan også se at fluktationen falder hvis intervallet bliver mindre eller hvis fordelingsvolumen bliver større.
Denne sammenhæng mellem dosisinterval og fluktation gør at man kan justere fluktationen, ved at justere dosis og interval mellem dosis sammenhængene, f.eks. halvere dosis og interval. Så bevares plasmakoncentrationen, men fluktationen bliver mindre. Fordelen ved en lav fluktation er færre bivirkninger pga. en høj -værdi og svigtende virkning pga. en for lav der ligger under den terapeutiske virkning. Ulemperne kan dog være de praktiske problemer der ligger med for ofte dosering.
Hvis man vil beregne gennemsnits steady state koncentrationen via van Rossums formel ved administration ved andet end intravaskulært, skal man inkludere absortionsfraktionen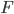 i formlen:
i formlen:
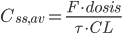
Arealet under plasmakoncentrations-tidskurven i et doseringsinterval ved steady state 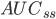 er lig med produktet af doseringsintervallet og gennemsnits steady-state koncentrationen. Vi får altså formlen:
er lig med produktet af doseringsintervallet og gennemsnits steady-state koncentrationen. Vi får altså formlen:
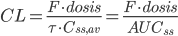
Hvis man gentager doseringerne og CL ikke ændrer sig væsentligt gælder der:
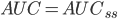
Støddosis
Hvis man skal behandle en akut sygdom, kan man ikke altid vente 5 halveringstider på at den terapeutiske dosis opnås. Man giver derfor en støddosis (kaldes også mætningsdosis eller loading dose).
Dosisafhængig kinetik
Dosisafhængig kinetik betyder at hvis man gentager en dosis, ændrer en farmakokinetisk parameter sig. Bl.a. kan man se ændring af clearence f.eks. pga. mætning af biotransformationen eller autoinduktion.
Sidst opdateret 31. maj 2023